
Scientists from the group found the naturally-occurring compound a hexakisphosphate (IP6) promotes both assembly and maturation of HIV-1. We discovered, in collaboration with other researchers, that HIV uses this small molecule to complete its function. This is a molecule that's extremely available in human cells and in other mammalian cells. HIV has evolved to make use of these small molecules present in our cells to essentially be infectious. Our study was published in the journal Nature in August 2018.
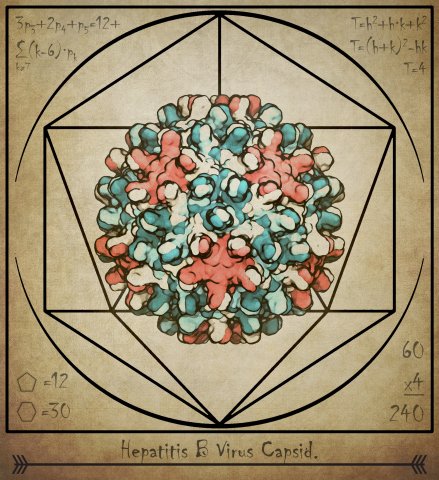
July 28 is World Hepatitis Day. The Biophysical Society spoke with us regarding our work on Hepatitis B. Hepatitis B is a potentially life-threatening liver infection caused by the hepatitis B virus (HBV). It can cause chronic infection and puts people at high risk of death from cirrhosis and liver cancer. According to the World Health Organization, an estimated 257 million people are living with hepatitis B virus infection.
More on the society's blog : Biophysics on world hepatitis day.
Hepatitis B virus (HBV) causes severe liver disease. Although a vaccine to prevent HBV is available, there is no cure to treat people who are already infected. Researchers aim to design new drugs against HBV that target its capsid, a protein shell at the core of the virus that encloses its genetic blueprint. During infection, the capsid drives delivery of the blueprint to the host cell nucleus, where it is used to generate new copies of the virus. While the structure of the HBV capsid had been previously determined, its motion, which underlies its role in HBV infection, had not been characterized at the atomic level until recently.
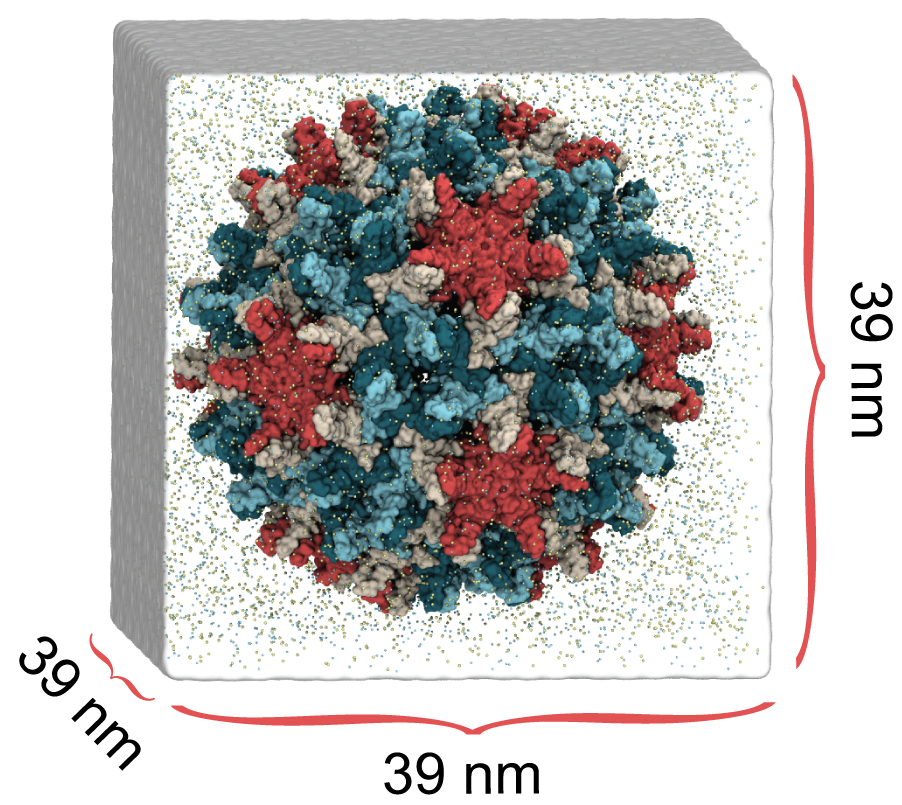
HBV capsid simulation system, 6M atoms
In a study published in eLife, we used molecular dynamics simulations on the microsecond timescale to observe the motion of the HBV capsid and its influence on surrounding water molecules and ions. We learned that the capsid is extremely flexible, and that this is likely the reason experimental microscopes have not been able to obtain high-resolution images of the capsid. We also learned that the capsid can distort asymmetrically, which may be important for the capsid to accommodate the asymmetric blueprint it encloses, or to squeeze through the nuclear pore into the host cell nucleus.
Finally, we learned that the capsid translocates ions across its surface through triangular-shaped pores, and that positively-charged ions translocate five times faster than negatively-charged ions. In the human body, the capsid proteins include positively-charged tails that expose themselves to the capsid surface, which signals the host cell to transport the capsid towards its nucleus. Our observations suggest that the tails are exposed through the capsid’s triangular pores, not through hexameric pores as previously thought, and that the capsid can control this exposure based on changes in tail charge that also occur during infection.
By revealing details about how the HBV capsid functions, our results imply new strategies to target the capsid with drugs. For example, drugs designed to rigidify the capsid could inhibit asymmetric distortion, and drugs designed to block triangular pores could inhibit cellular signaling. Importantly, our results also indicate that cryo-electron microscopy, which recently won the 2017 Nobel Prize in Chemistry, may be limited in reaching atomic resolution for biomolecules as flexible as the HBV capsid.

Maturation of HIV-1 particles encompasses a complex morphological transformation of Gag
via an orchestrated series of proteolytic cleavage events. A longstanding question concerns
the structure of the C-terminal region of CA and the peptide SP1 (CA–SP1), which represents
an intermediate during maturation of the HIV-1 virus. Employing enhanced sampling techniques in the Anton2 super computer researchers were able to observe the milisecond dynamics of the so-called SP1 domain. By integrating NMR, cryo-EM, and
molecular dynamics simulations, we show that SP1 acts as a maturation switch. In particular, we show that the SP1 peptide
exists in a dynamic helix–coil equilibrium, and that the addition of the maturation inhibitors
causes stabilization of a helical form of SP1. Moreover, the
maturation-arresting SP1 mutation T8I also induces helical structure in SP1 and further global
dynamical and conformational changes in CA. Overall, our results show that dynamics of CA
and SP1 are critical for orderly HIV-1 maturation and that small molecules can inhibit maturation by perturbing molecular motions. Results are published in the journal Nature Communications.

The list of World's most powerful supercomputers is out, and some of the computers employed by the group are listed in the Top500. Our research was higlighted as one of the key results from SuperComputers (see article here).

The Royal Swedish Academy of Sciences has decided to award the Nobel Prize in Chemistry 2017 to
Jacques Dubochet
University of Lausanne, Switzerland
Joachim Frank
Columbia University, New York, USA
and
Richard Henderson
MRC Laboratory of Molecular Biology, Cambridge, UK
"for developing cryo-electron microscopy for the high-resolution structure determination of biomolecules in solution"
Our lab continuosly develop novel techniques for cryoEM structure determination and refinement (see our list of Publications).
For a list of cryo-EM structures derived by our researchers please see :

Human dynamin–like, interferon-induced myxovirus resistance 2 (Mx2 or MxB) is a potent HIV-1 inhibitor. Antiviral activity requires both the amino-terminal region of MxB and protein oligomerization, each of which has eluded structural determination due to difficulties in protein preparation. We report that maltose binding protein–fused, full-length wild-type MxB purifies as oligomers and further self-assembles into helical arrays in physiological salt. Guanosine triphosphate (GTP), but not guanosine diphosphate, binding results in array disassembly, whereas subsequent GTP hydrolysis allows its reformation. Using cryo-electron microscopy (cryoEM), we determined the MxB assembly structure at 4.6 Å resolution, representing the first near-atomic resolution structure in the mammalian dynamin superfamily. The structure revealed previously described and novel MxB assembly interfaces. Mutational analyses demonstrated a critical role for one of the novel interfaces in HIV-1 restriction.
Human immunodeficiency virus type 1 (HIV-1) infection is highly dependent on its capsid. The capsid is a large container, made of ∼1,300 proteins with altogether 4 million atoms. Although the capsid proteins are all identical, they nevertheless arrange themselves into a largely asymmetric structure made of hexamers and pentamers. The large number of degrees of freedom and lack of symmetry pose a challenge to studying the chemical details of the HIV capsid. Simulations of over 64 million atoms for over 1 μs allow us to conduct a comprehensive study of the chemical–physical properties of an empty HIV-1 capsid, including its electrostatics, vibrational and acoustic properties, and the effects of solvent (ions and water) on the capsid. The simulations reveal critical details about the capsid with implications to biological function.
Juan Perilla and Jodi Hadden were allocated 582,000 NH for research focusing on virus capsids, the interactions of virus capsids with human factors and with antiviral drugs. Perilla says he and Hadden will use the allocation to study the effects of assembly inhibitor on the Hepatitis-B virus capsid and the HIV-1 capsid. “Blue Waters enables us to perform accurate, all-atom simulations of drug-compounds bound to the viral capsid and allows us to perform large-scale analysis of the results from the simulations,” said Pedilla.
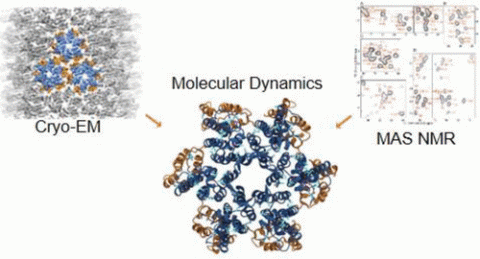
Single particle cryoEM has emerged as a powerful method for structure determination of proteins and complexes, complementing X-ray crystallography and NMR spectroscopy. Yet, for many systems, the resolution of cryoEM density map has been limited to 4–6 Å, which only allows for resolving bulky amino acids side chains, thus hindering accurate model building from the density map. On the other hand, experimental chemical shifts (CS) from solution and solid state MAS NMR spectra provide atomic level data for each amino acid within a molecule or a complex; however, structure determination of large complexes and assemblies based on NMR data alone remains challenging. In a recent publication, our group presented a novel integrated strategy to combine the highly complementary experimental data from cryoEM and NMR computationally by molecular dynamics simulations to derive an atomistic model, which is not attainable by either approach alone. We use the HIV-1 capsid protein (CA) C-terminal domain as well as the large capsid assembly to demonstrate the feasibility of this approach, termed NMR CS-biased cryoEM structure refinement.